4. CpG_density_gene_centered.py
4.1. Description
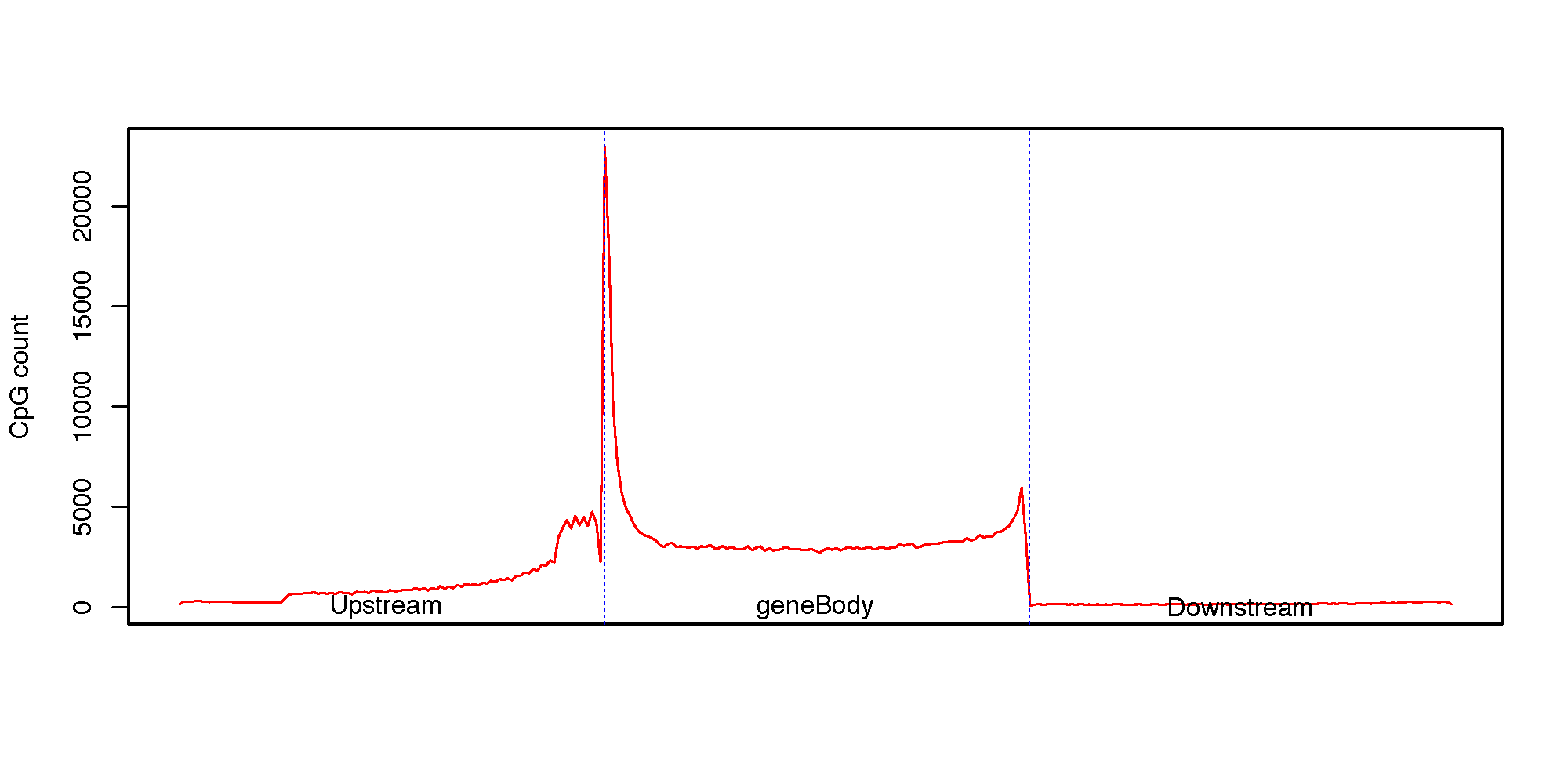
This program calculates the CpG density (count) profile over gene body as well as its up- down-stream regions. It is useful to visualize how CpGs are distributed around genes.
Specifically, the up-stream region, gene region (from TSS to TES) and down-stream region will be equally divided into 100 bins, then CpG count was aggregated over a total of 300 bins from 5’ to 3’ (upstream bins, gene bins, downstrem bins).
4.2. Options
- --version
show program’s version number and exit
- -h, --help
show this help message and exit
- -i INPUT_FILE, --input_file=INPUT_FILE
BED file specifying the C position. This BED file should have at least three columns (Chrom, ChromStart, ChromeEnd). Note: the first base in a chromosome is numbered 0. This file can be a regular text file or compressed file (.gz, .bz2).
- -r GENE_FILE, --refgene=GENE_FILE
Reference gene model in standard BED6+ format.
- -d DOWNSTREAM_SIZE, --downstream=DOWNSTREAM_SIZE
Maximum extension size from TES (transcription end site) to down-stream to define the “downstream intergenic region (DIR)”. Note: (1) The actual used DIR size can be smaller because the extending process could stop earlier if it reaches the boundary of another nearby gene. (2) If the actual used DIR size is smaller than cutoff defined by “-c/–SizeCut”, the gene will be skipped. default=2000 (bp)
- -u UPSTREAM_SIZE, --upstream=UPSTREAM_SIZE
Maximum extension size from TSS (transcription start site) to up-stream to define the “upstream intergenic region (UIR)”. Note: (1) The actual used UIR size can be smaller because the extending process could stop earlier if it reaches the boundary of another nearby gene. (2) If the actual used UIR size is smaller than cutoff defined by “-c/–SizeCut”, the gene will be skipped. default=2000 (bp)
- -c MINIMUM_SIZE, --SizeCut=MINIMUM_SIZE
The minimum gene size. Gene size is defined as the genomic size between TSS and TES, including both exons and introns. default=200 (bp)
- -o OUT_FILE, --output=OUT_FILE
The prefix of the output file.
4.3. Input files (examples)
4.4. Command
$ python3 CpG_density_gene_centered.py -r hg19.RefSeq.union.bed -i 850K_probe.hg19.bed3 -o CpG_density
@ 2020-03-11 14:57:10: Reading CpG file: "850K_probe.hg19.bed3"
@ 2020-03-11 14:57:14: Reading reference gene model: "hg19.RefSeq.union.bed"
@ 2020-03-11 14:57:14: Calculating CpG density ...
@ 2020-03-11 14:57:15: Wrting data to : "CpG_density.tsv"
@ 2020-03-11 14:57:15: Running R script to: 'CpG_density.r'
null device
1
4.5. Output files
CpG_density.tsv
CpG_density.r
CpG_density.pdf