Overview¶
CpGtools package provides a number of Python programs to annotate, QC, visualize, and analyze DNA methylation data generated from Illumina HumanMethylation450 BeadChip (450K) / MethylationEPIC BeadChip (850K) array or RRBS / WGBS.
These programs can be divided into three groups:
- CpG position analysis modules
- CpG signal analysis modules
- Differential CpG analysis modules
CpG position analysis modules¶
These modules are primarily used to analyze CpG’s genomic locations.
| Name | Description |
| CpG_aggregation.py | Aggregate proportion values of CpGs that located in give genomic regions (eg. CpG islands, promoters, exons, etc.). |
| CpG_anno_position.py | Add annotation information CpGs according to their genomic coordinates. |
| CpG_anno_probe.py | Add annotation information to 450K/850K probes. |
| CpG_density_gene_centered.py | Generate the CpG density (count) profile over gene body and the up/down-stream intergenic regions. |
| CpG_distrb_chrom.py | Calculate the distribution of CpG over chromosomes. |
| CpG_distrb_gene_centered.py | Calculate the distribution of CpG over gene-centered genomic regions. |
| CpG_distrb_region.py | Calculate the distribution of CpG over user-specified genomic regions. |
| CpG_logo.py | Generate a DNA motif logo and matrices for a given set of CpGs. |
| CpG_to_gene.py | Assign CpGs to their putative target genes. It uses the algorithm similar to GREAT. |
CpG signal analysis modules¶
These modules are primarily used to analyze CpG’s DNA methylation beta values
| Name | Description |
| beta_PCA.py | Perform PCA (principal component analysis) for samples. |
| beta_jitter_plot.py | Generate jitter plot (a.k.a. strip chart) and bean plot for each sample.” |
| beta_m_conversion.py | Convert Beta-value into M-value or vice versa. |
| beta_profile_gene_centered.py | Calculate the methylation profile (i.e., average beta value) for genomic regions around genes. |
| beta_profile_region.py | Calculate methylation profile (i.e. average beta value) around the user-specified genomic regions. |
| beta_stacked_barplot.py | Create stacked barplot for each sample. The stacked barplot showing the proportions of CpGs whose beta values are falling into [0,0.25], [0.25,0.5], [0.5,0.75],[0.75,1] |
| beta_stats.py | Summarize basic information on CpGs located in each genomic region. |
| beta_tSNE.py | Perform t-SNE (t-Distributed Stochastic Neighbor Embedding) analysis for samples. |
| beta_topN.py | Select the top N most variable CpGs (according to standard deviation) from the input file. |
| beta_trichotmize.py | Use Bayesian Gaussian Mixture model to trichotmize beta values into three status: ‘Un-methylated’,’Semi-methylated’, ‘Full-methylated’, and ‘unassigned’. |
| beta_UMAP.py | Perform UMAP (Uniform Manifold Approximation and Projection) for samples. |
Differential CpG analysis modules¶
These modules are primarily used to identify CpGs that are differentially methylated between groups
| Name | Description |
| dmc_Bayes.py | Differential CpG analysis using the Bayesian approach. (for 450K/850K data) |
| dmc_bb.py | Differential CpG analysis using the beta-binomial model. (for RRBS/WGBS count data) |
| dmc_fisher.py | Differential CpG analysis using Fisher’s Exact Test. (for RRBS/WGBS count data) |
| dmc_glm.py | Differential CpG analysis using the GLM generalized liner model. (for 450K/850K data) |
| dmc_logit.py | Differential CpG analysis using logistic regression model. (for RRBS/WGBS count data) |
| dmc_nonparametric.py | Differential CpG analysis using Mann-Whitney U test for two group comparison, and the Kruskal-Wallis H-test for multiple groups comparison. |
| dmc_ttest.py | Differential CpG analysis using T test. (for 450K/850K data) |
Installation¶
CpGtools are written in Python. Python3 (v3.5.x) is required to run all programs in CpGtools. Some programs also need R and R libraries to generate graphs and fit linear and beta-binomial models.
Prerequisites¶
Note: You need to install these tools if they are not available from your computer.
Python Dependencies¶
Note: You do NOT need to install these packages manually, as they will be automatically installed if you use pip3 to install CpGtools.
Install CpGtools using pip3 from PyPI or github¶
$ pip3 install cpgtools
or
$ pip3 install git+https://github.com/liguowang/cpgtools.git
Install CpGtools from source code¶
First, download the latest CpGtools, and then execute the following commands
$ tar zxf cpgtools-VERSION.tar.gz
$ cd cpgtools-VERSION
$ python3 setup.py install #install CpGtools to the default location
or
$ python3 setup.py install --root-/home/my_pylib/ #install CpGtools to user specified location
After the installation is completed, you probably need to setup up the environment variables (Below is only an example. Change according to your system configuration)
$ export PYTHONPATH-/home/my_pylib/python3.7/site-packages:$PYTHONPATH
Upgrade CpGtools¶
$ pip3 install cpgtools --upgrade
CpGtools Release history¶
Version 1.10.0¶
Add beta_UMAP.py on 09/24/2021
Version 1.0.8¶
Fix bug for beta_tSNE.py and beta_PCA.py when sample IDs are number.
Version 1.0.7¶
Add CpG_density_gene_centered.py on 03/11/2020
Version 1.0.2¶
- Add
beta_tSNE.pyon 07/15/2019 - This program performs t-SNE (t-Distributed Stochastic Neighbor Embedding) analysis for samples.
Version 1.0.1¶
- Add
CpG_anno_position.pyon 07/07/2019 - This program annotates CpG by its genomic position using prebuilt or user-provided annotation files.
Version 1.0.0¶
Initial release
Input file and data format¶
BED file¶
BED (Browser Extensible Data) format is commonly used to describe blocks of genome. The BED format consists of one line per feature, each containing 3-12 columns of data. It is 0-based (meaning the first base of a chromosome is numbered 0). It is s left-open, right-closed. For example, the bed entry “chr1 10 15” contains the 11-th, 12-th, 13-th, 14-th and 15-th bases of chromosome-1.
- BED12 file
- The standard BED file which has 12 fields. Each row in this file describes a gene or an array of disconnected genomic regions. Details are described here
- BED3 file
- Only has the first three required fields (chrom, chromStart, chromEnd). Each row is used to represent a single genomic region where “score” and “strand” are not necessary.
- BED3+ file
- Has at least three columns (chrom, chromStart, chromEnd). It could have other columns, but these additional columns will be ignored.
- BED6 file
- Has the first six fields (chrom, chromStart, chromEnd, name, score, strand). Each row is used to represent a single genomic region and their associated scores, or in cases where “strand” information is essential.
- BED6+ file
- Has at least six columns (chrom, chromStart, chromEnd, name, score, stand). It could have other columns, but these additional columns will be ignored.
Proportion values¶
In bisulfite sequencing (RRBS or WGBS), the methylation level of a particular CpG or region can be represented by a “proportion” value. We define the proportion value as a pair of integers separated by comma (“,”) with the first integer (m, 0 <- m <- n) representing “number of methylated reads” and the second integer (n, n >- 0) representing “number of total reads”. for example:
0,10 1,27 2,159 #Three proportions values indicated 3 hypo-methylated loci
7,7 17,19 30,34 #Three proportions values indicated 3 hyper-methylated loci
Beta values¶
The Beta-value is a value between 0 and 1, which can be interpreted as the approximation of the percentage of methylation for a given CpG or locus. One can convert proportion value into beta value, but not vice versa. In the equation below, C is the “probe intensity” or “read count” of methylated allele, while U is the “probe intensity” or “read count” of unmethylated allele.
M values¶
The M-value is calculated as the log2 ratio of the probe intensities (or read counts) of methylated allele versus unmethylated allele. In the equation below, C is the “probe intensity” or “read count” of methylated allele, while U is the “probe intensity” or “read count” of unmethylated allele. w is the offset or pseudo count added to both denominator and numerator to avoid unexpected big changes and performing log transformation on zeros.
Convert Beta value to M value or vice versa¶
The relationship between Beta-value and M-value is shown as equation and figure:

CpG_aggregation.py¶
Description¶
Aggregate proportion values of a list of CpGs that located in give genomic regions (eg. CpG islands, promoters, exons, etc.).
Example of input file
Chrom Start End score
chr1 100017748 100017749 3,10
chr1 100017769 100017770 0,10
chr1 100017853 100017854 16,21
Notes
Outlier CpG will be removed if the probability of observing its proportion value is less than p-cutoff. For example, if alpha set to 0.05, and there are 10 CpGs (n = 10) located in a particular genomic region, the p-cutoff of this genomic region is 0.005 (0.05/10). Supposing the total reads mapped to this region is 100, out of which 25 are methylated reads (i.e. regional methylation level beta = 25/100 = 0.25)
- The probability of observing CpG (3,10) is : pbinom(q=3, size=10, prob=0.25) = 0.7759
- The probability of observing CpG (0,10) is : pbinom(q=0, size=10, prob=0.25) = 0.05631
- The probability of observing CpG (16,21) is : pbinom(q=16, size=21, prob=0.25, lower.tail=F) = 1.19e-07 (outlier)
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input=INPUT_FILE Input CpG file in BED format. The first 3 columns contain “Chrom”, “Start”, and “End”. The 4th column contains proportion values. -a ALPHA_CUT, --alpha=ALPHA_CUT The chance of mistakingly assign a particular CpG as an outlier for each genomic region. default-0.05 -b BED_FILE, --bed=BED_FILE BED3+ file specifying the genomic regions. -o OUT_FILE, --output=OUT_FILE Prefix of the output file.
Input files (examples)¶
Command¶
$CpG_aggregation.py -b hg19.RefSeq.union.1Kpromoter.bed.gz -i test_03_RRBS.bed -o out
Output¶
chr1 567292 568293 3 0 93 3 0 93
chr1 713567 714568 6 0 100 6 0 100
chr1 762401 763402 7 0 110 7 0 110
chr1 762470 763471 10 0 158 10 0 158
chr1 854571 855572 2 12 16 2 12 16
chr1 860620 861621 16 91 232 16 91 232
chr1 894178 895179 12 151 229 41 506 735
Description
- Column1-3: Genome coordinates
- Column4-6: numbers of “CpG”, “aggregated methyl reads”, and “aggregate total reads” after outlier filtering
- Column7-9: numbers of “CpG”, “aggregated methyl reads”, and “aggregate total reads” before outlier filtering
CpG_anno_position.py¶
Description¶
This program adds annotation information to each CpG based on its genomic position.
Notes¶
- Input CpG (-i) and annotation (-a) BED files must have at least three columns, and must based on the same genome assembly version
- If multiple regions from the annotation BED file are overlapped with the same CpG site, their names will be concatenated together.
- Since the input (-i) is a regular BED foramt file, this module can be uesd to annotate any genomic regions of interest.
Pre-computed datasets¶
- hg19_ENCODE_338TF_130Cell_E3.bed.gz (File size = 108.2 MB)
- Transcription factor (TF) binding sites identified from ChIP-seq experiments performed by the ENCODE project. Peaks from 1264 experiments representing 338 transcription factors in 130 cell types are combined (N = 10,560,472). BED format file was downloaded from the UCSC Tabel Browser.
- hg19_ENCODE_DNaseI_125Cells_V3.bed.gz (File size = 24.3 MB)
- DNase I hypersensitivity sites identified from ENCODE DNase-seq experiments. Peaks from 125 cell types are combined (N = 1,867,665). BED format file was downloaded from the UCSC Tabel Browser.
- hg19_ENCODE_chromHMM_states_9Cells.merge.bed.gz (File size = 32.7 MB)
- Chromatin State Segmentation by chromHMM from ENCODE. Chromatin states across 9 cell types (GM12878, H1-hESC, K562, HepG2, HUVEC, HMEC, HSMM, NHEK, NHLF) were learned by integrating 9 factors (CTCF, H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me2, H3K4me3, H3K9ac, H4K20me1 ) plus input. A total of 15 states were identified, include: State-1 (Active Promoter), state-2 (Weak Promoter), state-3 (Inactive/poised Promoter), state-4 and 5 (Strong enhancer), state-6 and 7 (Weak/poised enhancer), state-8 (insulator), state-9 (Transcriptional transition), state-10 (Transcriptional elongation), state-11 (Weak transcribed), state-12 (Polycomb-repressed), state-13 (Heterochromatin or low signal), state-14 and 15 (Repetitive/Copy Number Variation). The Original chromatin state BED file was downloaded from the UCSC Tabel Browser.
- hg19_FANTOM_enhancers_phase_1_and_2.bed.gz
- PHANTOM5 human permissive enhancers downloaded from here.
- hg19_ENCODE_H3K4me1_11_cellLines_ChIP.bed.gz (File size = 12.2 MB)
- H3K4me1 (marker of active and primed enhancer) peaks identified from ENCODE histone ChIP-seq experiments. Peaks from 11 cell types (GM12878, H1-hESC, HMEC, HSMM, HUVEC, HeLaS3, HepG2, K562, Monocytes-CD14+_RO01746, NHEK, NHLF) are combined (N = 1,435,550)
- hg19_ENCODE_H3K4me3_11_cellLines_ChIP.bed.gz (File size = 4.5 MB)
- H3K4me3 (marker of promoter) peaks identified from ENCODE histone ChIP-seq experiments. Peaks from 11 cell types (GM12878, H1-hESC, HMEC, HSMM, HUVEC, HeLaS3, HepG2, K562, Monocytes-CD14+_RO01746, NHEK, NHLF) are combined (N = 525,824)
- hg19_ENCODE_H3K27ac_11_cellLines_ChIP.bed.gz (File size = 5.7 MB)
- H3K27ac (marker of active enhancer) peaks identified from ENCODE histone ChIP-seq experiments. Peaks from 11 cell types (GM12878, H1-hESC, HMEC, HSMM, HUVEC, HeLaS3, HepG2, K562, Monocytes-CD14+_RO01746, NHEK, NHLF) are combined (N = 665,650)
These BED files were lifted over to hg38/GRCh38 using CrossMap. The hg38-based annotation files are available from here
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Input CpG file in BED3+ format. -a ANNO_FILE, --annotation=ANNO_FILE Input annotation file in BED3+ format. -w WINDOW_SIZE, --window=WINDOW_SIZE Size of window centering on the middle-point of each genomic region defined in the annotation BED file (i.e., window_size*0.5 will be extended to up- and down-stream from the middle point of each genomic region). default=100 -o OUT_FILE, --output=OUT_FILE The prefix of the output file. -l, --header If True, the first row of input CpG file is header. default=False
Input files (examples)¶
Command¶
$CpG_anno_position.py -l -a hg19_ENCODE_338TF_130Cell_E3.bed.gz -i test_01.hg19.bed6 -o output
Output files¶
- output.anno.txt
$ head -5 output.anno.txt
#Chrom Start End Name Beta Strand hg19_ENCODE_338TF_130Cell_E3.bed
chr1 10847 10848 cg26928153 0.8965 + N/A
chr1 10849 10850 cg16269199 0.7915 + N/A
chr1 15864 15865 cg13869341 0.9325 + N/A
chr1 534241 534242 cg24669183 0.7941 + FOXA2,MNT
CpG_anno_probe.py¶
Description¶
This program adds comprehensive annotation information to each 450K/850K array probe ID. It will add 17 columns to the original input data file. These 17 columns include (from left to right):
| Header Name | Description |
| hg19_pos | The genomic position of the CpG on human genome assembly hg19 (or GRCh37) |
| hg38_pos | The genomic position of the CpG on human genome assembly hg38 (or GRCh38). |
| strand | Strand of the CpG. Value - “R” (reverse strand) or “F” (forward strand). |
| geneSymbol | Genes the CpG has been assigned to. “N/A” indicates no genes were found. This is retrieved from the Illumina MethylationEpic v1.0 B4 manifest file. |
| CpGisland | The CpG island (CGI) that overlaps with this CpG. “N/A” indicates no CGIs were found. |
| with_450K | Boolean indicating whether this CpG probe is also included in 450K. “0” - No, “1”- Yes. |
| SNP_ID | SNPs (rsID) that are close to this CpG. Multiple SNPs are separated by “;”. “N/A” indicates no SNPs were found. |
| SNP_distance | The nucleotide distances between SNPs and the CpG. |
| SNP_MAF | The minor allele frequencies (MAF) of SNPs. |
| Cross_Reactive | Boolean (“0” - No, “1”- Yes) indicating whether this CpG could be affected by cross-hybridization or underlying genetic variation as reported by this paper. |
| ENCODE_TF_ChIP | Transcription factor (TF) binding sites identified from ChIP-seq experiments performed by the ENCODE project. Peaks from 1264 experiments representing 338 transcription factors in 130 cell types are combined (N = 10,560,472). BED format file was downloaded from the UCSC Tabel Browser, and a detailed description is provided here. |
| ENCODE_DNaseI | DNase I hypersensitivity sites identified from ENCODE DNase-seq experiments. Peaks from 125 cell types are combined (N - 1,867,665). BED format file was downloaded from the UCSC Table Browser, and a detailed description is provided here. |
| ENCODE_H3K27ac_ChIP | H3K27ac peaks identified from ENCODE histone ChIP-seq experiments. Peaks from 11 cell types (GM12878, H1-hESC, HMEC, HSMM, HUVEC, HeLaS3, HepG2, K562, Monocytes-CD14+_RO01746, NHEK, NHLF) are combined (N = 665,650) |
| ENCODE_H3K4me1_ChIP | H3K4me1 peaks identified from ENCODE histone ChIP-seq experiments. Peaks from 11 cell types (GM12878, H1-hESC, HMEC, HSMM, HUVEC, HeLaS3, HepG2, K562, Monocytes-CD14+_RO01746, NHEK, NHLF) are combined (N = 1,435,550) |
| ENCODE_H3K4me3_ChIP | H3K4me3 peaks identified from ENCODE histone ChIP-seq experiments. Peaks from 11 cell types (GM12878, H1-hESC, HMEC, HSMM, HUVEC, HeLaS3, HepG2, K562, Monocytes-CD14+_RO01746, NHEK, NHLF) are combined (N = 525,824) |
| ENCODE_chromHMM | Chromatin State Segmentation by chromHMM from ENCODE. Chromatin states across 9 cell types (GM12878, H1-hESC, K562, HepG2, HUVEC, HMEC, HSMM, NHEK, NHLF) were learned by computationally by integrating 9 factors (CTCF, H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me2, H3K4me3, H3K9ac, H4K20me1 ) plus input. A total of 15 states were identified, include: State-1 (Active Promoter), state-2 (Weak Promoter), state-3 (Inactive/poised Promoter), state-4 and 5 (Strong enhancer), state-6 and 7 (Weak/poised enhancer), state-8 (insulator), state-9 (Transcriptional transition), state-10 (Transcriptional elongation), state-11 (Weak transcribed), state-12 (Polycomb-repressed), state-13 (Heterochromatin or low signal), state-14 and 15 (Repetitive/Copy Number Variation). Orignal chromatin state BED file was downloaded from UCSC Table Browser, and detailed description is provided here. |
| FANTOM_enhancer | PHANTOM5 human enhancers downloaded from here. |
Notes¶
- For peaks identified from ENCODE ChIP-seq and DNase-seq (ENCODE_TF_ChIP, ENCODE_H3K27ac_ChIP, ENCODE_H3K4me1_ChIP, ENCODE_H3K4me3_ChIP, and ENCODE_DNaseI), we require the probe must be located in the 100 bp window centered on the middle of the peak.
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Input data file (Tab-separated) with a certain column containing 450K/850K array CpG IDs. This file can be a regular text file or compressed file (.gz, .bz2). -a ANNO_FILE, --annotation=ANNO_FILE Annotation file. This file can be a regular text file or compressed file (.gz, .bz2). -o OUT_FILE, --output=OUT_FILE Prefix of the output file. -p PROBE_COL, --probe_column=PROBE_COL The number specifying which column contains probe IDs. Note: the column index starts with 0. default-0. -l, --header Input data file has a header row.
Input files (examples)¶
Command¶
# probe IDs are located in the 4th column (-p 3)
$CpG_anno_probe.py -p 3 -l -a MethylationEPIC_CpGtools.tsv -i test_01.hg19.bed6 -o output
or (take gzipped files as input)
$CpG_anno_probe.py -p 3 -l -a MethylationEPIC_CpGtools.tsv.gz -i test_01.hg19.bed6.gz -o output
@ 2019-06-28 09:12:41: Read annotation file "../epic/MethylationEPIC_CpGtools.tsv" ...
@ 2019-06-28 09:12:52: Add annotation information to "test_01.hg19.bed6" ...
Output files¶
- output.anno.txt
CpG_density_gene_centered.py¶
Description¶
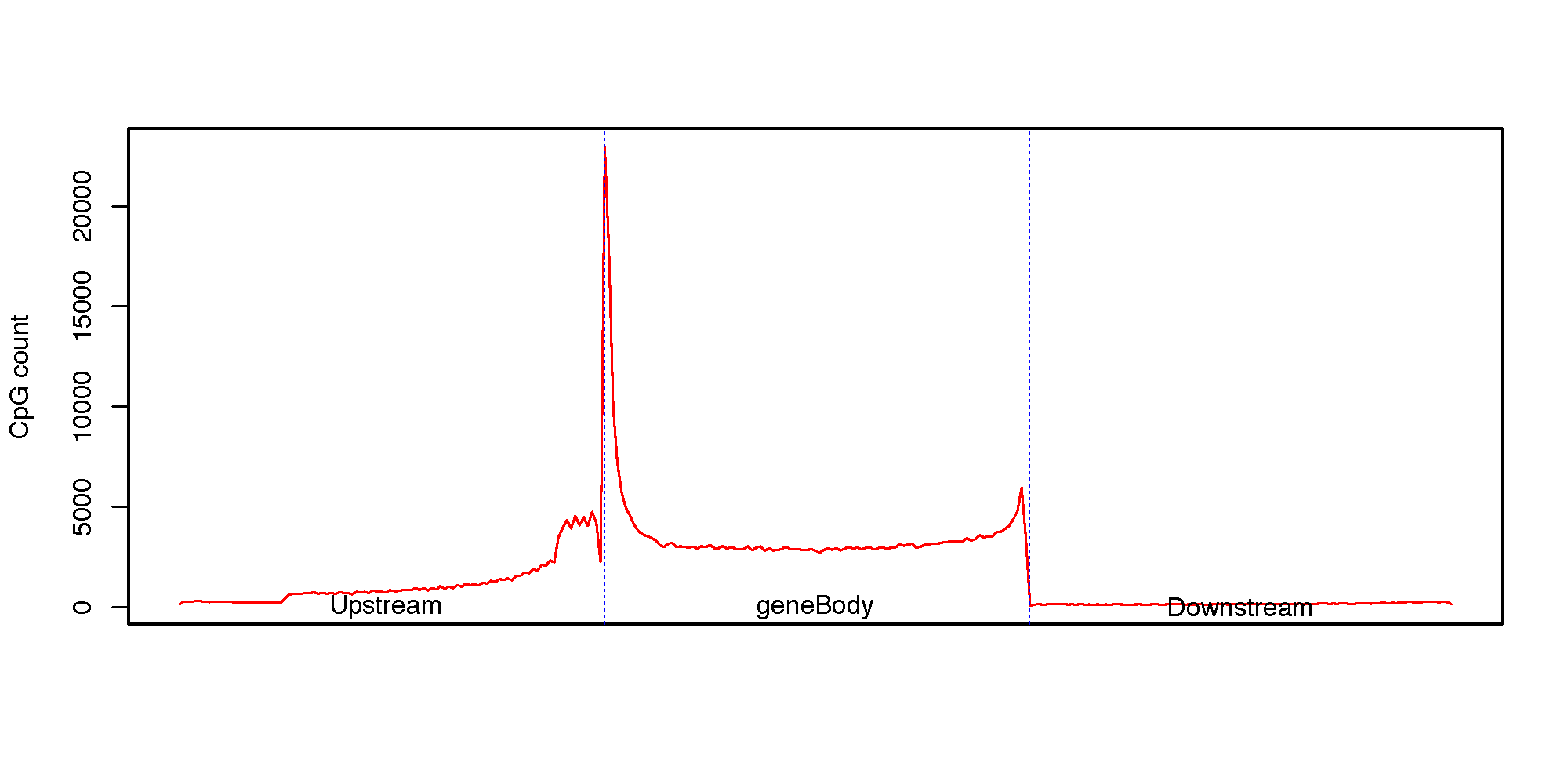
This program calculates the CpG density (count) profile over gene body as well as its up- down-stream regions. It is useful to visualize how CpGs are distributed around genes.
Specifically, the up-stream region, gene region (from TSS to TES) and down-stream region will be equally divided into 100 bins, then CpG count was aggregated over a total of 300 bins from 5’ to 3’ (upstream bins, gene bins, downstrem bins).
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE BED file specifying the C position. This BED file should have at least three columns (Chrom, ChromStart, ChromeEnd). Note: the first base in a chromosome is numbered 0. This file can be a regular text file or compressed file (.gz, .bz2). -r GENE_FILE, --refgene=GENE_FILE Reference gene model in standard BED6+ format. -d DOWNSTREAM_SIZE, --downstream=DOWNSTREAM_SIZE Maximum extension size from TES (transcription end site) to down-stream to define the “downstream intergenic region (DIR)”. Note: (1) The actual used DIR size can be smaller because the extending process could stop earlier if it reaches the boundary of another nearby gene. (2) If the actual used DIR size is smaller than cutoff defined by “-c/–SizeCut”, the gene will be skipped. default=2000 (bp) -u UPSTREAM_SIZE, --upstream=UPSTREAM_SIZE Maximum extension size from TSS (transcription start site) to up-stream to define the “upstream intergenic region (UIR)”. Note: (1) The actual used UIR size can be smaller because the extending process could stop earlier if it reaches the boundary of another nearby gene. (2) If the actual used UIR size is smaller than cutoff defined by “-c/–SizeCut”, the gene will be skipped. default=2000 (bp) -c MINIMUM_SIZE, --SizeCut=MINIMUM_SIZE The minimum gene size. Gene size is defined as the genomic size between TSS and TES, including both exons and introns. default=200 (bp) -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$ python3 CpG_density_gene_centered.py -r hg19.RefSeq.union.bed -i 850K_probe.hg19.bed3 -o CpG_density
@ 2020-03-11 14:57:10: Reading CpG file: "850K_probe.hg19.bed3"
@ 2020-03-11 14:57:14: Reading reference gene model: "hg19.RefSeq.union.bed"
@ 2020-03-11 14:57:14: Calculating CpG density ...
@ 2020-03-11 14:57:15: Wrting data to : "CpG_density.tsv"
@ 2020-03-11 14:57:15: Running R script to: 'CpG_density.r'
null device
1

CpG_distrb_chrom.py¶
Description¶
This program calculates the distribution of CpG over chromosomes
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILES, --input_files=INPUT_FILES Input CpG file(s) in BED3+ format. Multiple BED files should be separated by “,” (eg: “-i file_1.bed,file_2.bed,file_3.bed”). BED file can be a regular text file or compressed file (.gz, .bz2). The barplot figures will NOT be generated if you provide more than 12 samples (bed files). [required] -n FILE_NAMES, --names=FILE_NAMES Shorter and meaningful names to label samples. Should be separated by “,” and match CpG BED files in number. If not provided, basenames of CpG BED files will be used to label samples. [optional] -s CHROM_SIZE, --chrom-size=CHROM_SIZE Chromosome size file. Tab or space separated text file with two columns: the first column is chromosome name/ID, the second column is chromosome size. This file will determine: (1) which chromosomes are included in the final bar plots, so do NOT include ‘unplaced’, ‘alternative’ contigs in this file. (2) The order of chromosomes in the final bar plots. [required] -o OUT_FILE, --output=OUT_FILE The prefix of the output file. [required]
Input files (examples)¶
Command¶
$ chrom_distribution.py -i 450K_probe.hg19.bed3.gz,850K_probe.hg19.bed3.gz -n 450K,850K \
-s hg19.chrom.sizes -o chromDist
Output files
- chromDist.txt
- chromDist.r
- chromDist.CpG_total.pdf
- chromDist.CpG_percent.pdf
- chromDist.CpG_perMb.pdf
Total CpG count per chromosome

CpG percent on each chromosome (normalized to total CpGs)

CpG per Mb (normalized to chromosome size)

CpG_distrb_gene_centered.py¶
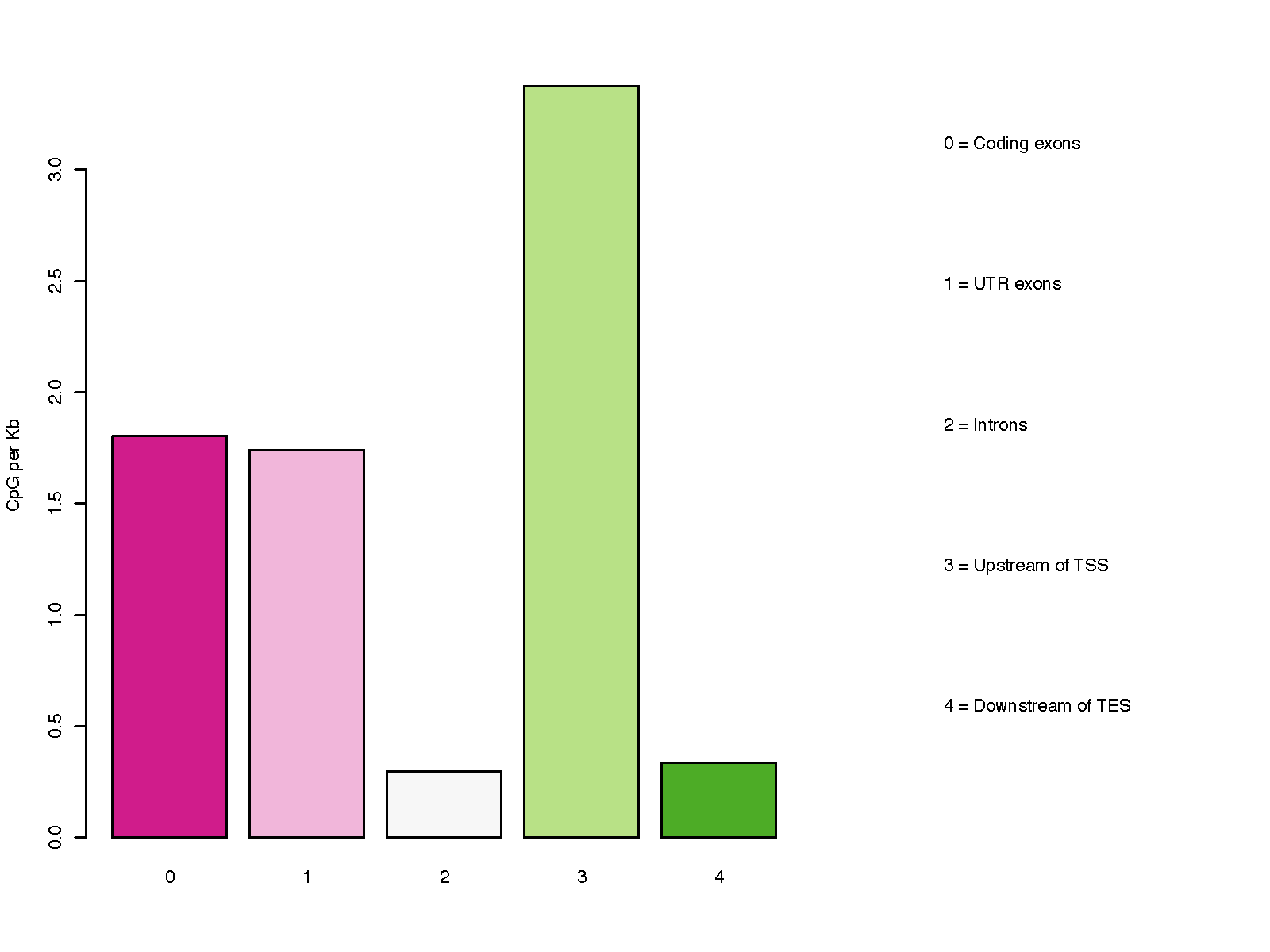
Description¶
This program calculates the distribution of CpG over gene-centered genomic regions including ‘Coding exons’, ‘UTR exons’, ‘Introns’, ‘ Upstream intergenic regions’, and ‘Downsteam intergenic regions’.
Notes
Please note, a particular genomic region can be assigned to different groups listed above, because most genes have multiple transcripts, and different genes could overlap on the genome. For example, an exon of gene A could be located in an intron of gene B. To address this issue, we define the priority order as below:
- Coding exons
- UTR exons
- Introns
- Upstream intergenic regions
- Downstream intergenic regions
Higher-priority group override the low-priority group. For example, if a certain part of an intron is overlapped with an exon of other transcripts/genes, the overlapped part will be considered as exon (i.e., removed from intron) since “exon” has higher priority.
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE BED file specifying the C position. This BED file should have at least three columns (Chrom, ChromStart, ChromeEnd). Note: the first base in a chromosome is numbered 0. This file can be a regular text file or compressed file (.gz, .bz2). -r GENE_FILE, --refgene=GENE_FILE Reference gene model in standard BED-12 format (https://genome.ucsc.edu/FAQ/FAQformat.html#format1). -d DOWNSTREAM_SIZE, --downstream=DOWNSTREAM_SIZE Size of down-stream intergenic region w.r.t. TES (transcription end site). default=2000 (bp) -u UPSTREAM_SIZE, --upstream=UPSTREAM_SIZE Size of up-stream intergenic region w.r.t. TSS (transcription start site). default=2000 (bp) -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$ CpG_distrb_gene_centered.py -i 850K_probe.hg19.bed3.gz -r hg19.RefSeq.union.bed.gz -o geneDist

CpG_distrb_region.py¶
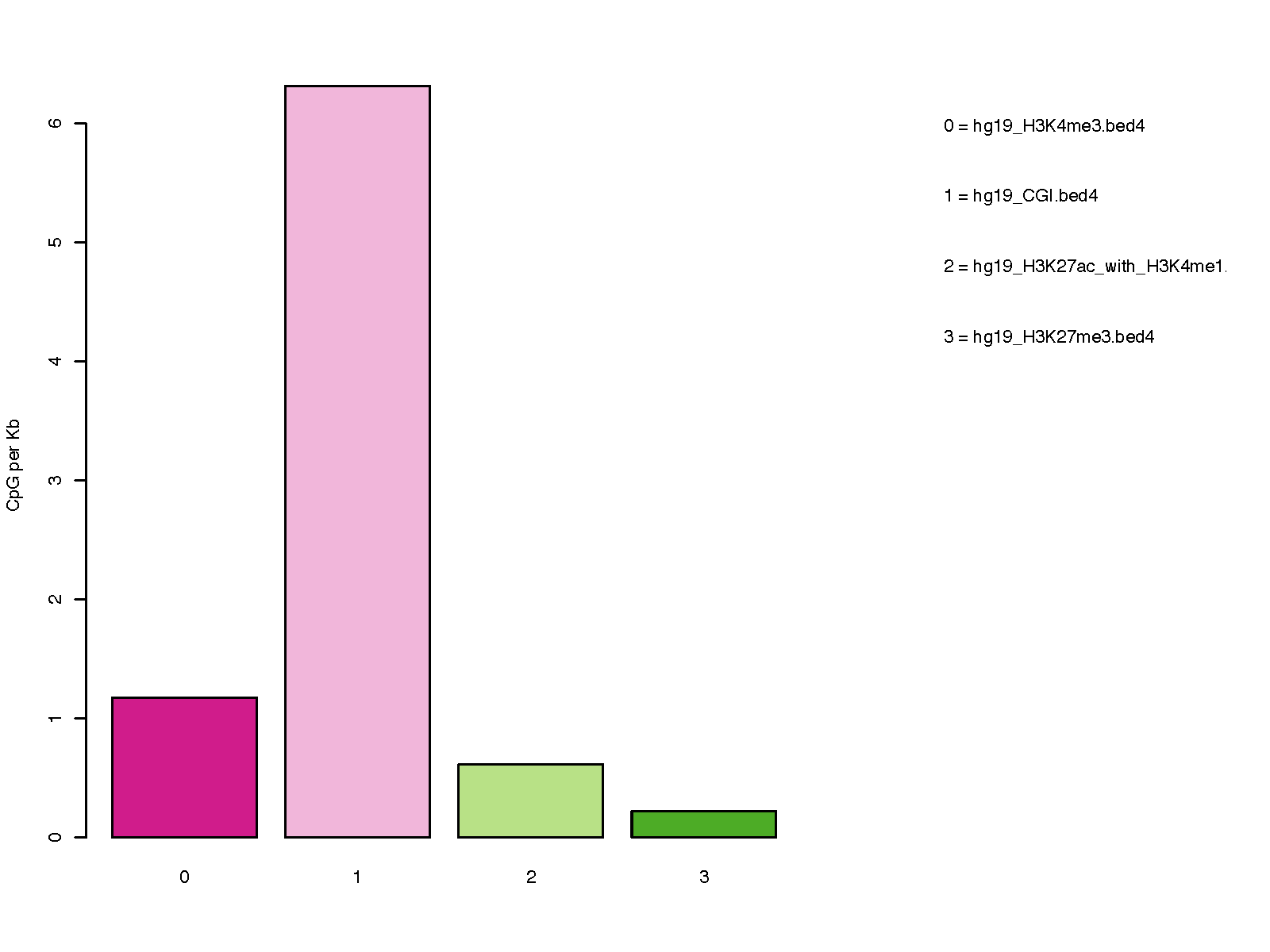
Description¶
This program calculates the distribution of CpG over user-specified genomic regions.
Notes
- A maximum of ten BED files (define ten different genomic regions) can be analyzed together.
- The order of BED files is important (i.e., considered as “priority order”). Overlapped genomic regions will be kept in the BED file with the highest priority and removed from BED files of lower priorities. For example, users provided 3 BED files via “-i promoters.bed,enhancers.bed,intergenic.bed”, then if an enhancer region is overlapped with promoters, the overlapped part will be removed from “enhancers.bed”.
- BED files can be regular or compressed by ‘gzip’ or ‘bz’.
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i CPG_FILE, --cpg=CPG_FILE BED file specifying the C position. This BED file should have at least three columns (Chrom, ChromStart, ChromeEnd). Note: the first base in a chromosome is numbered 0. This file can be a regular text file or compressed file (.gz, .bz2). -b BED_FILES, --bed=BED_FILES List of comma separated BED files specifying the genomic regions. -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
- 850K_probe.hg19.bed3.gz Input bed file of 850K probe
- hg19_CGI.bed4 CpG islands
- hg19_H3K4me3.bed4 Promoters
- hg19_H3K27ac_with_H3K4me1.bed4 Bivalent promoters
- hg19_H3K27me3.bed4 Heterochromatin regions
Command¶
# check the distribution of 850K probes in 4 genomic regions (CpG islands, Promoters,
# Bivalent promoters, and Heterochromatin regions)
$CpG_distrb_region.py -i 850K_probe.hg19.bed3.gz -b hg19_H3K4me3.bed4,hg19_CGI.bed4,\
hg19_H3K27ac_with_H3K4me1.bed4,hg19_H3K27me3.bed4 -o regionDist

CpG_logo.py¶
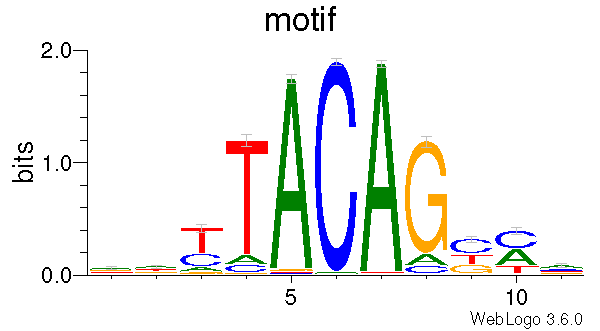
Description¶
This program generates a DNA motif logo for a given set of CpGs. To answer the question of “what is the genomic context for a given list of CpGs ?”. This program first extracts genomic sequences around C position, and then generate motif matrices include:
- position frequency matrix (PFM)
- position probability matrix (PPM)
- position weight matrix (PWM)
- MEME format matrix
- Jaspar format matrix
It also generates motif logo using weblogo
Notes
- input BED file must have strand information.
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE BED file specifying the C position. This BED file should have at least six columns (Chrom, ChromStart, ChromeEnd, name, score, strand). Note: Must provide correct strand information. This file can be a regular text file or compressed file (.gz, .bz2). -r GENOME_FILE, --refgenome=GENOME_FILE Reference genome seqeunces in FASTA format. Must be indexed using the samtools “faidx” command. -e EXTEND_SIZE, --extend=EXTEND_SIZE Number of bases extended to up- and down-stream. default=5 (bp) -n MOTIF_NAME, --name=MOTIF_NAME Motif name. default=motif -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
- Human reference genome sequences in FASTA format: hg19.fa.gz and hg38.fa.gz
- 450_CH.hg19.bed.gz
Command¶
$CpG_logo.py -i 450_CH.hg19.bed.gz -r hg19.fa -o 450_CH
Output files¶
- 450_CH.logo.fa
- 450_CH.logo.jaspar
- 450_CH.logo.meme
- 450_CH.logo.pfm
- 450_CH.logo.ppm
- 450_CH.logo.pwm
- 450_CH.logo.logo.pdf
CpG_to_gene.py¶
Description¶
This program annotates CpGs by assigning them to their putative target genes. It follows the “Basal plus extension rules” used by GREAT.
Basal regulatory domain is a user-defined genomic region around the TSS (transcription start site). By default, from TSS upstream 5 Kb to TSS downstream 1 Kb is considered as the gene’s basal regulatory domain. When defining a gene’s basal regulatory domain, the other nearby genes are ignored (which means different genes’ basal regulatory domain can be overlapped.)
Extended regulatory domain is a genomic region that is further extended from basal regulatory domain in both directions to the nearest gene’s basal regulatory domain but no more than the maximum extension (specified by ‘-e’, default - 1000 kb) in one direction. In other words, the “extension” stops when it reaches other genes’ “basal regulatory domain” or the extension limit, whichever comes first.
Basal regulatory domain and Extended regulatory domain are illustrated in below diagram.

Notes
- Which genes are assigned to a particular CpG largely depends on gene annotation. A “conservative” gene model (such as Refseq curated protein-coding genes) is recommended.
- In the refgene file, multiple isoforms should be merged into a single gene.
Description¶
This program annotates CpGs by assigning them to their putative target genes. Follows the “Basel plus extension” rules used by GREAT(http://great.stanford.edu/public/html/index.php)
- Basal regulatory domain: is a user-defined genomic region around the TSS (transcription start site). By default, from TSS upstream 5kb to TSS downstream 1Kb is considered as the gene’s basal regulatory domain. When defining a gene’s “basal regulatory domain”, the other nearby genes will be ignored (which means different genes’ basal regulatory domains can be overlapped.)
- Extended regulatory domain: The gene regulatory domain is extended in both directions to the nearest gene’s “basal regulatory domain” but no more than the maximum extension (default = 1000 kb) in one direction.
Notes¶
- Which genes are assigned to a particular CpG largely depends on gene annotation. A “conservative” gene model (such as Refseq curated protein coding genes) is recommended.
- In the gene model, multiple isoforms should be merged into a single gene.
Options¶
- Options:
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input-file=INPUT_FILE BED3+ file specifying the C position. BED3+ file could be a regular text file or compressed file (.gz, .bz2). [required] -r GENE_FILE, --refgene=GENE_FILE Reference gene model in BED12 format (https://genome.ucsc.edu/FAQ/FAQformat.html#format1). “One gene one transcript” is recommended. Since most genes have multiple transcripts; one can collapse multiple transcripts of the same gene into a single super transcript or select the canonical transcript. -u BASAL_UP_SIZE, --basal-up=BASAL_UP_SIZE Size of extension to upstream of TSS (used to define gene’s “basal regulatory domain”). default=5000 (bp) -d BASAL_DOWN_SIZE, --basal-down=BASAL_DOWN_SIZE Size of extension to downstream of TSS (used to define gene’s basal regulatory domain). default=1000 (bp) -e EXTENSION_SIZE, --extension=EXTENSION_SIZE Size of extension to both up- and down-stream of TSS (used to define gene’s “extended regulatory domain”). default=1000000 (bp) -o OUT_FILE, --output=OUT_FILE Prefix of the output file. Two additional columns will be appended to the original BED file with the last column indicating “genes whose extended regulatory domain are overlapped with the CpG”, the 2nd last column indicating “genes whose basal regulatory domain are overlapped with the CpG”. [required]
Input files (examples)¶
Command¶
$ CpG_to_gene.py -i 850K_probe.hg19.bed3.gz -r hg19.RefSeq.union.bed.gz -o output
Output files¶
- output.associated_genes.txt
$ head output.associated_genes.txt
#The last column contains genes whose extended regulatory domain are overlapped with the CpG
#The 2nd last column contains genes whose basal regulatory domain are overlapped with the CpG
#"//" indicates no genes are found
chr1 10524 10525 DDX11L1 //
chr1 10847 10848 DDX11L1 //
chr1 10849 10850 DDX11L1 //
chr1 15864 15865 // MIR6859-1;DDX11L1
chr1 18826 18827 MIR6859-1 //
chr1 29406 29407 WASH7P;MIR1302-2 //
chr1 29424 29425 WASH7P;MIR1302-2 //
...
beta_PCA.py¶
Description¶
This program performs PCA (principal component analysis) for samples.
Example of input data file
ID Sample_01 Sample_02 Sample_03 Sample_04
cg_001 0.831035 0.878022 0.794427 0.880911
cg_002 0.249544 0.209949 0.234294 0.236680
cg_003 0.845065 0.843957 0.840184 0.824286
...
Example of input group file
Sample,Group
Sample_01,normal
Sample_02,normal
Sample_03,tumor
Sample_04,tumo
...
Notes
- Rows with missing values will be removed
- Beta values will be standardized into z scores
- Only the first two components will be visualized
- Variance% explained by each component will be printed to screen
- Options:
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Tab-separated data frame file containing beta values with the 1st row containing sample IDs and the 1st column containing CpG IDs. -g GROUP_FILE, --group=GROUP_FILE Comma-separated group file defining the biological groups of each sample. Different groups will be colored differently in the PCA plot. Supports a maximum of 20 groups. -n N_COMPONENTS, --ncomponent=N_COMPONENTS Number of components. default=2 -l, --label If True, sample ids will be added underneath the data point. default=False -c PLOT_CHAR, --char=PLOT_CHAR Ploting character: 1 = ‘dot’, 2 = ‘circle’. default=1 -a PLOT_ALPHA, --alpha=PLOT_ALPHA Opacity of dots. default=0.5 -x LEGEND_LOCATION, --loc=LEGEND_LOCATION Location of legend panel: 1 = ‘topright’, 2 = ‘bottomright’, 3 = ‘bottomleft’, 4 = ‘topleft’. default=1 -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$beta_PCA.py -i cirrHCV_vs_normal.data.tsv -g cirrHCV_vs_normal.grp.csv -o HCV_vs_normal

beta_UMAP.py¶
Description¶
This program performs UMAP (Uniform Manifold Approximation and Projection) non-linear dimension reduction.
Example of input data file
ID Sample_01 Sample_02 Sample_03 Sample_04
cg_001 0.831035 0.878022 0.794427 0.880911
cg_002 0.249544 0.209949 0.234294 0.236680
cg_003 0.845065 0.843957 0.840184 0.824286
...
Example of input group file
Sample,Group
Sample_01,normal
Sample_02,normal
Sample_03,tumor
Sample_04,tumo
...
Notes
- Rows with missing values will be removed
- Beta values will be standardized into z scores
- Only the first two components will be visualized
- Options:
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Tab-separated data frame file containing beta values with the 1st row containing sample IDs and the 1st column containing CpG IDs. -g GROUP_FILE, --group=GROUP_FILE Comma-separated group file defining the biological groups of each sample. Different groups will be colored differently in the 2-dimensional plot. Supports a maximum of 20 groups. -n N_COMPONENTS, --ncomponent=N_COMPONENTS Number of components. default=2 --nneighbors=N_NEIGHBORS This parameter controls the size of the local neighborhood UMAP will look at when attempting to learn the manifold structure of the data. Low values of ‘–nneighbors’ will force UMAP to concentrate on local structure, while large values will push UMAP to look at larger neighborhoods of each point when estimating the manifold structure of the data. Choose a value from [2, 200]. default=15 --min-dist=MIN_DISTANCE This parameter controls how tightly UMAP is allowed to pack points together. Choose a value from [0, 1). default=0.2 -l, --label If True, sample ids will be added underneath the data point. default=False -c PLOT_CHAR, --char=PLOT_CHAR Ploting character: 1 = ‘dot’, 2 = ‘circle’. default=1 -a PLOT_ALPHA, --alpha=PLOT_ALPHA Opacity of dots. default=0.5 -x LEGEND_LOCATION, --loc=LEGEND_LOCATION Location of legend panel: 1 = ‘topright’, 2 = ‘bottomright’, 3 = ‘bottomleft’, 4 = ‘topleft’. default=1 -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$beta_UMAP.py -i cirrHCV_vs_normal.data.tsv -g cirrHCV_vs_normal.grp.csv -o cirrHCV_vs_normal -l

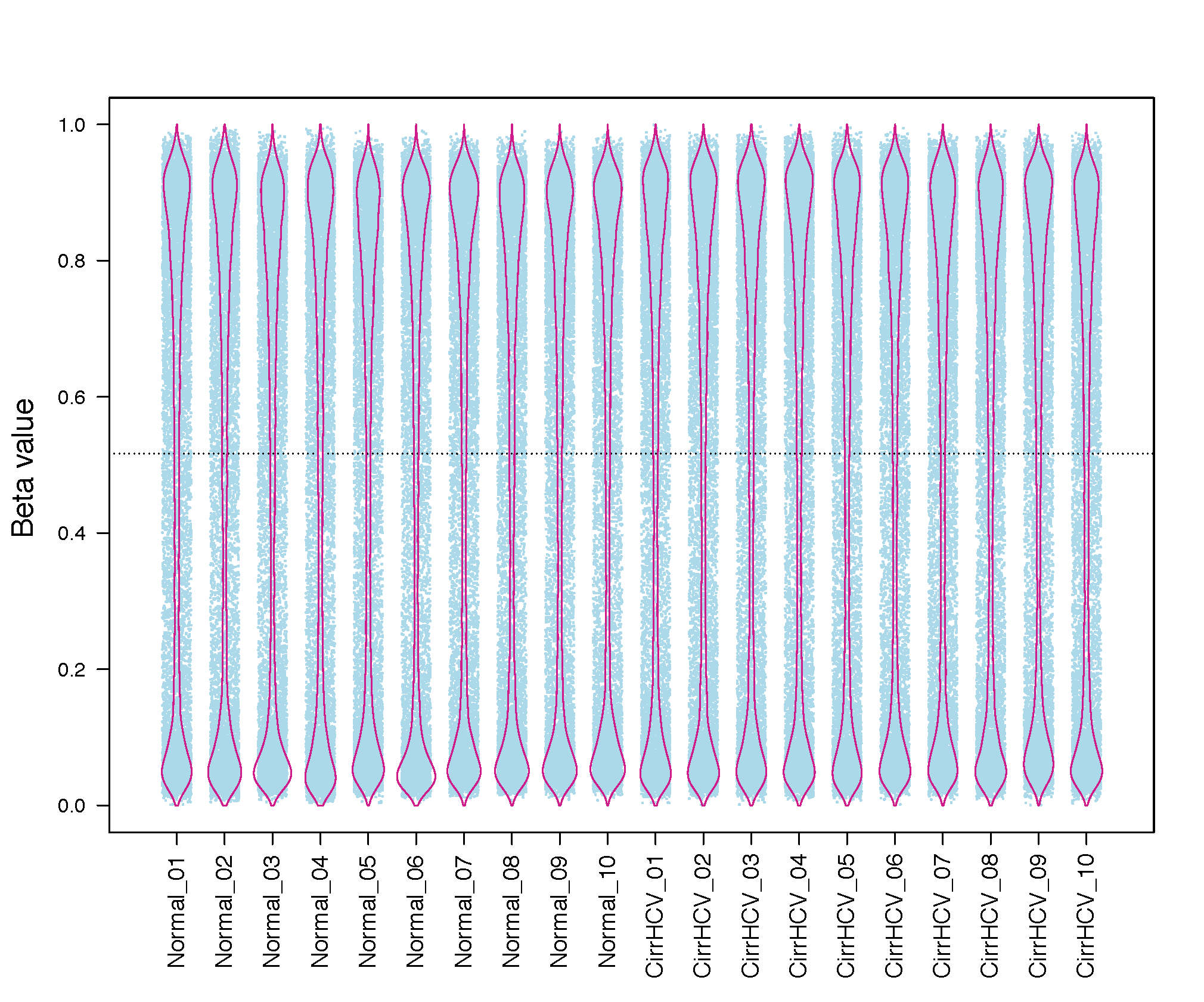
beta_jitter_plot.py¶
Description¶
This program generates jitter plot (a.k.a. strip chart) and bean plot for each sample (column)
Example of input
CpG_ID Sample_01 Sample_02 Sample_03 Sample_04
cg_001 0.831035 0.878022 0.794427 0.880911
cg_002 0.249544 0.209949 0.234294 0.236680
cg_003 0.845065 0.843957 0.840184 0.824286
Notes
- User must install the beanplot R library.
- Please name your sample IDs (such as “Sample_01”, “Sample_02” in the above example) using only “letters” [a-z, A-Z], “numbers” [0-9], and “_”; and your sample ID must start with a letter.
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Tab-separated data frame file containing beta values with the 1st row containing sample IDs and the 1st column containing CpG IDs. -f FRACTION, --fraction=FRACTION The fraction of total data points (CpGs) used to generate jitter plot. Decrease this number if the jitter plot is over-crowded. default=0.5 -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$beta_jitterPlot.py -f 1 -i test_05_TwoGroup.tsv.gz -o Jitter

beta_m_conversion.py¶
Description¶
Convert Beta-value into M-value or vice vers
Example of input (beta)
CpG_ID Sample_01 Sample_02 Sample_03 Sample_04 cg_001 0.831035 0.878022 0.794427 0.880911 cg_002 0.249544 0.209949 0.234294 0.236680 cg_003 0.845065 0.843957 0.840184 0.824286
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Tab-separated data frame file containing beta values with the 1st row containing sample IDs and the 1st column containing CpG IDs. This file can be a regular text file or compressed file (.gz, .bz2). -d DATA_TYPE, --dtype=DATA_TYPE Input data type either “Beta” or “M”. -o OUT_FILE, --output=OUT_FILE The output file.
Input file (example)¶
Command¶
$ beta_m_conversion.py -i test_08.tsv -d Beta -o test_08_M.tsv
Output¶
$ head -5 test_08_M.tsv
cg_ID TCGA-BC-A10Q TCGA-BC-A10R TCGA-BC-A10S TCGA-BC-A10T TCGA-BC-A10U
cg00000029 -0.9127840676229807 -0.6635535075463712 -0.9389653708375745 -1.1786876012968779 -0.6217264255944122
cg00000165 -2.4833534763405667 -2.3330364850204406 -2.858145170950326 -2.914508967160336 -2.3645896606652745
cg00000236 2.478873972561897 3.0777336083377693 2.6760378499862143 3.04301970048709 2.7096166677505145
cg00000289 0.9943771370790748 0.13339998728363872 0.5981994318909333 1.2402989291699527 1.432741941887314
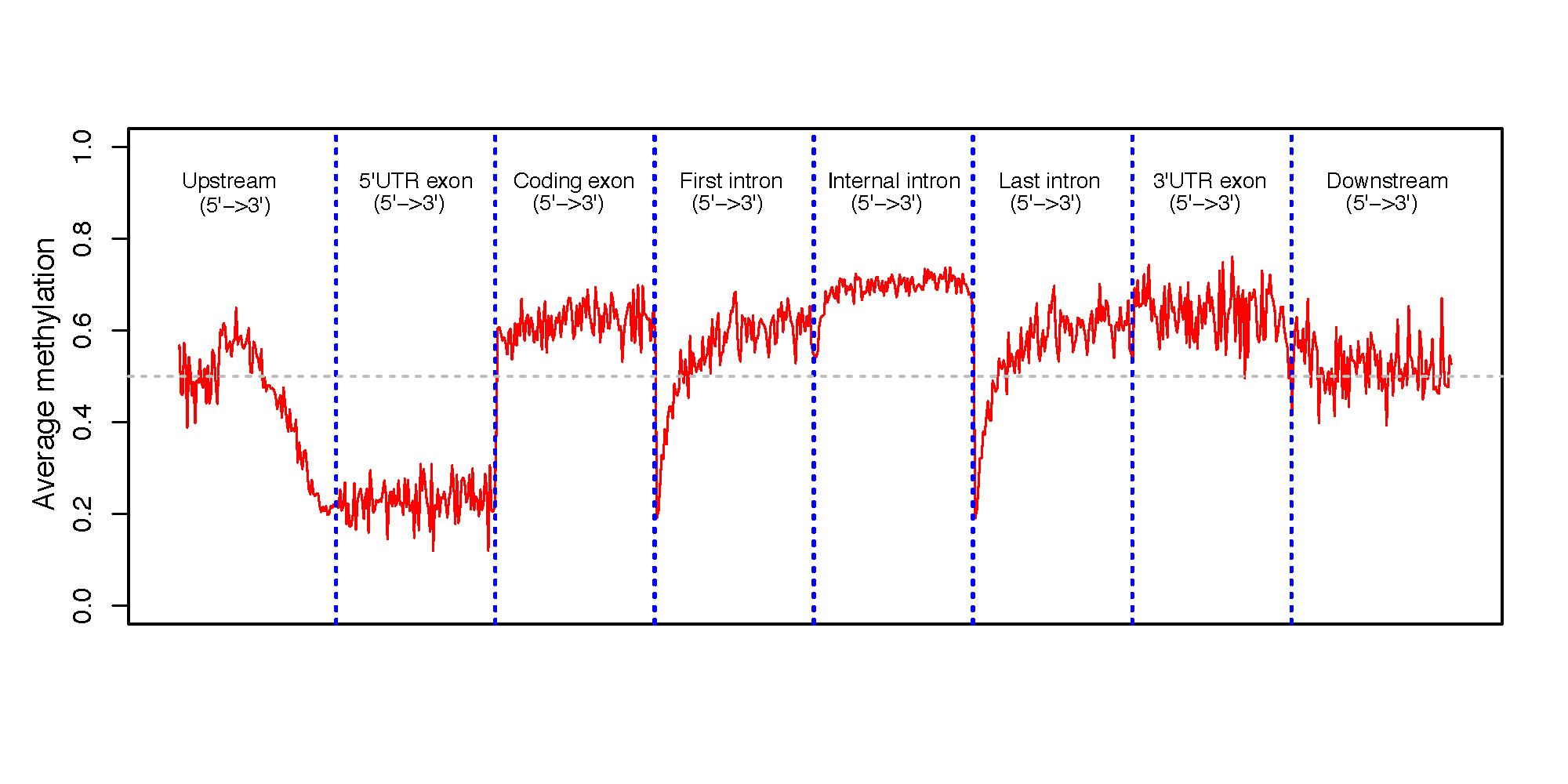
beta_profile_gene_centered.py¶
Description¶
This program calculates the methylation profile (i.e., average beta value) for genomic regions around genes. These genomic regions include:
- 5’UTR exon
- CDS exon
- 3’UTR exon,
- first intron
- internal intron
- last intron
- up-stream intergenic
- down-stream intergenic
Example of input (BED6+)
chr22 44021512 44021513 cg24055475 0.9231 -
chr13 111568382 111568383 cg06540715 0.1071 +
chr20 44033594 44033595 cg21482942 0.6122 -
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE BED6+ file specifying the C position. This BED file should have at least 6 columns (Chrom, ChromStart, ChromeEnd, Name, Beta_value, Strand). BED6+ file can be a regular text file or compressed file (.gz, .bz2). -r GENE_FILE, --refgene=GENE_FILE Reference gene model in standard BED12 format (https://genome.ucsc.edu/FAQ/FAQformat.html#format1). “Strand” column must exist in order to decide 5’ and 3’ UTRs, up- and down-stream intergenic regions. -d DOWNSTREAM_SIZE, --downstream=DOWNSTREAM_SIZE Size of down-stream genomic region added to gene. default=2000 (bp) -u UPSTREAM_SIZE, --upstream=UPSTREAM_SIZE Size of up-stream genomic region added to gene. default=2000 (bp) -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Command¶
$beta_profile_gene_centered.py -i test_02.bed6.gz -r hg19.RefSeq.union.bed.gz -o gene_profile

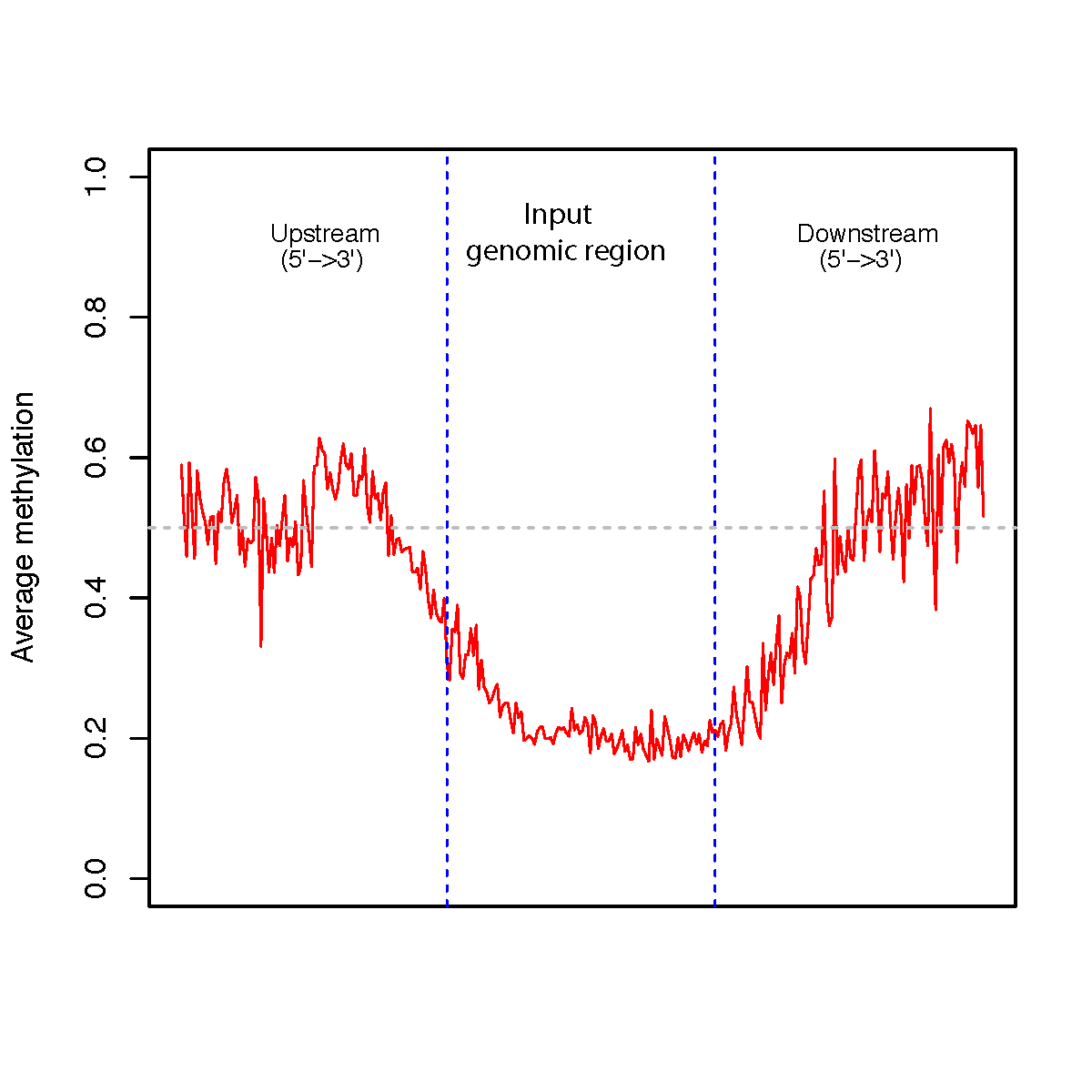
beta_profile_region.py¶
Description¶
This program calculates methylation profile (i.e. average beta value) around the user-specified genomic regions.
Example of input
# BED6 format (INPUT_FILE)
chr22 44021512 44021513 cg24055475 0.9231 -
chr13 111568382 111568383 cg06540715 0.1071 +
chr20 44033594 44033595 cg21482942 0.6122 -
# BED3 format (REGION_FILE)
chr1 15864 15865
chr1 18826 18827
chr1 29406 29407
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE BED6+ file specifying the C position. This BED file should have at least six columns (Chrom, ChromStart, ChromeEnd, Name, Beta_value, Strand). BED6+ file can be a regular text file or compressed file (.gz, .bz2). -r REGION_FILE, --region=REGION_FILE BED3+ file of genomic regions. This BED file should have at least three columns (Chrom, ChromStart, ChromeEnd). If the 6-th column does not exist, all regions will be considered as on “+” strand. -d DOWNSTREAM_SIZE, --downstream=DOWNSTREAM_SIZE Size of extension to downstream. default=2000 (bp) -u UPSTREAM_SIZE, --upstream=UPSTREAM_SIZE Size of extension to upstream. default=2000 (bp) -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$beta_profile_region.py -r hg19.RefSeq.union.1Kpromoter.bed.gz -i test_02.bed6.gz -o region_profile

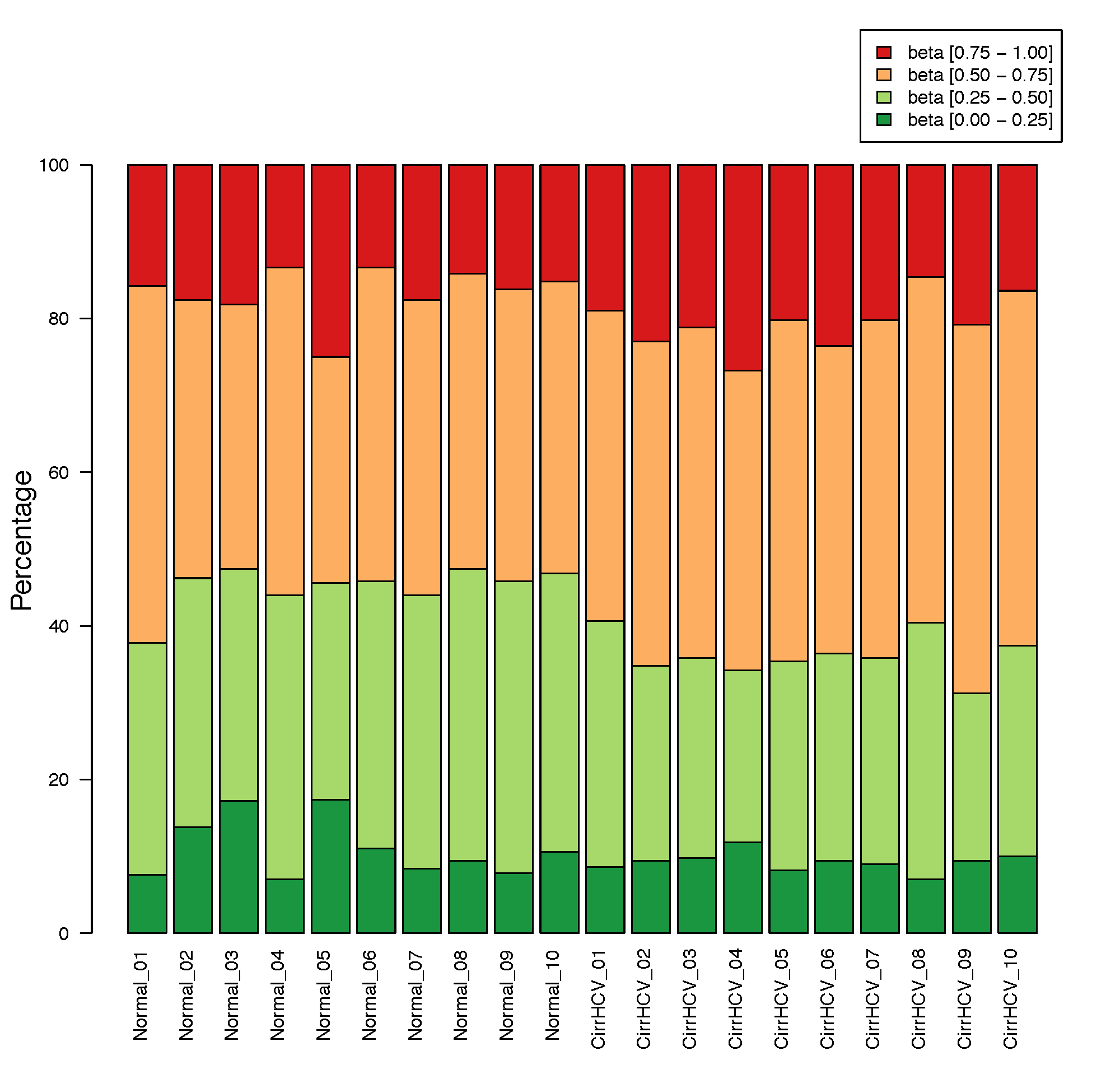
beta_stacked_barplot.py¶
Description¶
This program creates stacked barplot for each sample. The stacked barplot showing the proportions of CpGs whose beta values are falling into these four ranges:
- [0.00, 0.25] #first quantile
- [0.25, 0.50] #second quantile
- [0.50, 0.75] #third quantile
- [0.75, 1.00] #forth quantile
Example of input file
CpG_ID Sample_01 Sample_02 Sample_03 Sample_04
cg_001 0.831035 0.878022 0.794427 0.880911
cg_002 0.249544 0.209949 0.234294 0.236680
Notes
- Please name your sample IDs (such as “Sample_01”, “Sample_02” in the above example) using only “letters” [a-z, A-Z], “numbers” [0-9], and “_”; and your sample ID must start with a letter.
Options¶
- Options:
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Data frame file containing beta values with the 1st row containing sample IDs and the 1st column containing CpG IDs. -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$beta_stacked_barplot.py -i cirrHCV_vs_normal.data.tsv -o stacked_bar

beta_stats.py¶
Description¶
This program gives basic information on CpGs located in each genomic region. It adds 6 columns to the input BED file:
- Number of CpGs detected in the genomic region
- Min methylation level
- Max methylation level
- Average methylation level across all CpGs
- Median methylation level across all CpGs
- Standard deviation
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE BED6+ file specifying the C position. This BED file should have at least six columns (Chrom, ChromStart, ChromeEnd, Name, Beta_value, Strand). Note: the first base in a chromosome is numbered 0. This file can be a regular text file or compressed file (.gz, .bz2) -r REGION_FILE, --region=REGION_FILE BED3+ file of genomic regions. This BED file should have at least 3 columns (Chrom, ChromStart, ChromeEnd). -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$beta_stats.py -r hg19.RefSeq.union.1Kpromoter.bed.gz -i test_02.bed6.gz -o region_stats
Output files¶
- region_stats.txt
beta_tSNE.py¶
Description¶
This program performs t-SNE (t-Distributed Stochastic Neighbor Embedding) analysis for samples.
Example of input data file
ID Sample_01 Sample_02 Sample_03 Sample_04
cg_001 0.831035 0.878022 0.794427 0.880911
cg_002 0.249544 0.209949 0.234294 0.236680
cg_003 0.845065 0.843957 0.840184 0.824286
...
Example of input group file
Sample,Group
Sample_01,normal
Sample_02,normal
Sample_03,tumor
Sample_04,tumo
...
Notes
- Rows with missing values will be removed
- Beta values will be standardized into z scores
- Only the first two components will be visualized
- Different perplexity values can result in significantly different results
- Even with same data and save parameters, different run might give you (slightly) different result. It is perfectly fine to run t-SNE a number of times (with the same data and parameters), and to select the visualization with the lowest value of the objective function as your final visualization.
Options:
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Tab-separated data frame file containing beta values with the 1st row containing sample IDs and the 1st column containing CpG IDs. -g GROUP_FILE, --group=GROUP_FILE Comma-separated group file defining the biological groups of each sample. Different groups will be colored differently in the t-SNE plot. Supports a maximum of 20 groups. -p PERPLEXITY_VALUE, --perplexity=PERPLEXITY_VALUE This is a tunable parameter of t-SNE, and has a profound effect on the resulting 2D map. Consider selecting a value between 5 and 50, and the selected value should be smaller than the number of samples (i.e., number of points on the t-SNE 2D map). Default = 5 -n N_COMPONENTS, --ncomponent=N_COMPONENTS Number of components. default=2 --n_iter=N_ITERATIONS The maximum number of iterations for the optimization. Should be at least 250. default=5000 --learning_rate=LEARNING_RATE The learning rate for t-SNE is usually in the range [10.0, 1000.0]. If the learning rate is too high, the data may look like a ‘ball’ with any point approximately equidistant from its nearest neighbors. If the learning rate is too low, most points may look compressed in a dense cloud with few outliers. If the cost function gets stuck in a bad local minimum increasing the learning rate may help. default=200.0 -l, --label If True, sample ids will be added underneath the data point. default=False -c PLOT_CHAR, --char=PLOT_CHAR Ploting character: 1 = ‘dot’, 2 = ‘circle’. default=1 -a PLOT_ALPHA, --alpha=PLOT_ALPHA Opacity of dots. default=0.5 -x LEGEND_LOCATION, --loc=LEGEND_LOCATION Location of legend panel: 1 = ‘topright’, 2 = ‘bottomright’, 3 = ‘bottomleft’, 4 = ‘topleft’. default=1 -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$beta_tSNE.py -i cirrHCV_vs_normal.data.tsv -g cirrHCV_vs_normal.grp.csv -o HCV_vs_normal

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
beta_topN.py¶
Description¶
This program picks the top N rows (according to standard deviation) from the input file. The resulting file can be used for clustering and PCA analysis.
Example of input
CpG_ID Sample_01 Sample_02 Sample_03 Sample_04
cg_001 0.831035 0.878022 0.794427 0.880911
cg_002 0.249544 0.209949 0.234294 0.236680
cg_003 0.845065 0.843957 0.840184 0.824286
Options¶
- Options:
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Tab-separated data frame file containing beta values with the 1st row containing sample IDs and the 1st column containing CpG IDs. -c CPG_COUNT, --count=CPG_COUNT Number of most variable CpGs (ranked by standard deviation) to keep. default=1000 -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$beta_topN.py -i test_05_TwoGroup.tsv.gz -c 500 -o test_05_TwoGroup
Output file¶
- test_05_TwoGroup.sortedStdev.tsv
- test_05_TwoGroup.sortedStdev.topN.tsv
beta_trichotmize.py¶
Description¶
Rather than using a hard threshold to call “methylated” or “unmethylated” CpGs or regions, this program uses a probability approach (Bayesian Gaussian Mixture model) to trichotmize beta values into three status:
- Un-methylated : labeled as “0” in the result file
- Both the homologous chromosomes (i.e. The maternal and paternal chromosomes) are unmethylated.
- Semi-methylated : labeled as “1” in the result file
- Only one of the homologous chromosomes is methylated. This is also called allele-specific methylation or imprinting. Note: semi-methylation here is different from hemimethylation, which refers to “one of two (complementary) strands is methylated”.
- Full-methylated : labeled as “2” in the result file
- Both the homologous chromosomes (i.e., The maternal and paternal chromosomes) are methylated.
- unassigned : labeled as “-1” in the result file
- CpGs failed to assigned to the three categories above.
Algorithm¶
As described above, in somatic cells, most CpGs can be grouped into 3 categories including “Un-methylated”, “Semi-methylated (imprinted)” and “Full-methylated”. Therefore, the Beta distribution of CpGs can be considered as the mixture of 3 Gaussian distributions (i.e. components). beta_trichotmize.py first estimates the parameters (mu1, mu2, mu3) and (s1, s2, s3) of the 3 components using expectation–maximization (EM) algorithm, then it calculates the posterior probabilities ( p0, p1, and p2) of each component given the beta value of a CpG.
- p0
- the probability that the CpG belongs to un-methylated component.
- p1
- the probability that the CpG belongs to semi-methylated component.
- p2
- the probability that the CpG belongs to full-methylated component.
The classification will be made using rules:
if p0 == max(p0, p1, p2):
un-methylated
elif p2 == max(p0, p1, p2):
full-methylated
elif p1 == max(p0, p1, p2):
if p1 >= prob_cutoff:
semi-methylated
else:
unknown/unassigned
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Input plain text file containing beta values with the 1st row containing sample IDs (must be unique) and the 1st column containing probe IDs (must be unique). -c PROB_CUTOFF, --prob-cut=PROB_CUTOFF Probability cutoff to assign a probe into “semi- methylated” class. default=0.99 -r, --report If True, generates “summary_report.txt” file. default=False -s RANDOM_STATE, --seed=RANDOM_STATE The seed used by the random number generator. default=99
Input files (examples)¶
Command¶
$beta_trichotmize.py -i test_05_TwoGroup.tsv -r
Output files¶
- .results.txt for each sample
- summary_report.txt
$ head CirrHCV_01.results.txt
#Prob_of_0: Probability of CpG belonging to un-methylation group
#Prob_of_1: Probability of CpG belonging to semi-methylation group
#Prob_of_2: Probability of CpG belonging to full-methylation group
#Assigned_lable: -1 = 'unassigned', 0 = 'un-methylation', 1 = 'semi-methylation', 2 = 'full-methylation'
Probe_ID Beta_value Prob_of_1 Prob_of_0 Prob_of_2 Assigned_lable
cg00000109 0.8776539440000001 0.05562534330044164 3.673659573888142e-93 0.9443746566995583 2
cg00000165 0.239308082 0.999222373166152 0.0007776268338481155 1.3380168478281785e-21 1
cg00000236 0.8951333909999999 0.052142920095512614 3.5462722261710256e-97 0.9478570799044873 2
cg00000292 0.783661275 0.22215555206863843 1.46921724055509e-72 0.7778444479313614 2
cg00000321 0.319783971 0.9999999909047641 9.09523558157906e-09 1.4703488768311725e-16 1
$ cat summary_report.txt
#means of components
Subject_ID Unmethyl SemiMethyl Methyl
CirrHCV_01 0.0705891104729628 0.4949428535816466 0.8694861885234295
CirrHCV_02 0.06775600800214297 0.5018649959502874 0.8731195740516192
CirrHCV_03 0.07063205540113326 0.49795240946021674 0.8730234341971185
...
#Weights of components
Subject_ID Unmethyl SemiMethyl Methyl
CirrHCV_01 0.27231055290074735 0.35186129618859385 0.3758281509106588
CirrHCV_02 0.2623073658620772 0.36736674559925425 0.37032588853866855
CirrHCV_03 0.2659211619015646 0.3563058727320757 0.37777296536635974
...
#Converge status and n_iter
Subject_ID Converged n_iter
CirrHCV_01 True 35
CirrHCV_02 True 37
CirrHCV_03 True 34
Below histogram and piechart showed the proportion of CpGs assigned to “Un-methylated”, “Semi-methylated” and “Full-methylated”.

dmc_Bayes.py¶
Description¶
Different from statistical testing, this program tries to estimates “how different the means between the two groups are” using the Bayesian approach. An MCMC is used to estimate the “means”, “difference of means”, “95% HDI (highest posterior density interval)”, and the posterior probability that the HDI does NOT include “0”.
It is similar to John Kruschke’s BEST algorithm (Bayesian Estimation Supersedes T test)
Notes
- This program is much slower than T-test due to MCMC (Markov chain Monte Carlo) step. Running it with multiple threads is highly recommended.
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Data file containing beta values with the 1st row containing sample IDs (must be unique) and the 1st column containing CpG positions or probe IDs (must be unique). Except for the 1st row and 1st column, any non-numerical values will be considered as “missing values” and ignored. This file can be a regular text file or compressed file (.gz, .bz2). -g GROUP_FILE, --group=GROUP_FILE Group file defining the biological group of each sample. It is a comma-separated 2 columns file with the 1st column containing sample IDs, and the 2nd column containing group IDs. It must have a header row. Sample IDs should match to the “Data file”. Note: Only for two group comparison. -n N_ITER, --niter=N_ITER Iteration times when using MCMC Metropolis-Hastings’s agorithm to draw samples from the posterior distribution. default=5000 -b N_BURN, --burnin=N_BURN Number of simulated samples to discard. Thes initial samples are usually not completely valid because the Markov Chain has not stabilized to the stationary distribution. default=500. -p N_PROCESS, --processor=N_PROCESS The number of processes. default=1 -s SEED, --seed=SEED The seed used by the random number generator. default=99 -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$ dmc_Bayes.py -i test_05_TwoGroup.tsv.gz -g test_05_TwoGroup.grp.csv.gz -p 10 -o dmc_output
Output files¶
- dmc_output.bayes.tsv: this file consists of 6 columns:
- ID : CpG ID
- mu1 : Mean methylation level estimated from group1
- mu2 : Mean methylation level estimated from gropu2
- mu_diff : Difference between mu1 and mu2
- mu_diff (95% HDI) : 95% of “High Density Interval” of mu_diff. The HDI indicates which points of distribution are most credible. This interval spans 95% of mu_diff’s distribution.
- The probability that mu1 and mu2 are different.
$head -10 dmc_output.bayes.tsv
ID mu1 mu2 mu_diff mu_diff (95% HDI) Probability
cg00001099 0.775209 0.795404 -0.020196 (-0.065148,0.023974) 0.811024
cg00000363 0.610565 0.469523 0.141042 (0.030769,0.232965) 0.994665
cg00000884 0.845973 0.873761 -0.027787 (-0.051976,-0.004398) 0.984882
cg00000714 0.190868 0.199233 -0.008365 (-0.030071,0.014006) 0.816141
cg00000957 0.772905 0.827528 -0.054623 (-0.092116,-0.016465) 0.995327
cg00000292 0.748394 0.766326 -0.017932 (-0.051286,0.012583) 0.889729
cg00000807 0.729162 0.683732 0.045430 (-0.001523,0.086588) 0.981551
cg00000721 0.935903 0.935080 0.000823 (-0.013210,0.018628) 0.508686
cg00000948 0.898609 0.897536 0.001073 (-0.020663,0.026813) 0.518238
mu1 and mu2 can be considered as significantly different if the 95% HDI does NOT include zero.
dmc_bb.py¶
Description¶
This program performs differential CpG analysis using the beta-binomial model. It allows for covariant analysis.
Notes - You must install R package aod before running this program.
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Data file containing methylation proportions (represented by “methyl_count,total_count”, eg. “20,30”) with the 1st row containing sample IDs (must be unique) and the 1st column containing CpG positions or probe IDs (must be unique). This file can be a regular text file or compressed file (.gz, .bz2) -g GROUP_FILE, --group=GROUP_FILE Group file defining the biological groups of each sample as well as other covariables such as gender, age. The first variable is grouping variable (must be categorical), all the other variables are considered as covariates (can be categorical or continuous). Sample IDs should match to the “Data file”. -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files¶
Command¶
$ python3 ../bin/dmc_bb.py -i test_04_TwoGroup.tsv.gz -g test_04_TwoGroup.grp.csv -o OUT_bb
dmc_fisher.py¶
Description¶
This program performs differential CpG analysis using Fisher exact test on proportion value. It applies to two sample comparison with no biological/technical replicates. If biological/ technical replicates are provided, methyl reads and total reads of all replicates will be merged (i.e. ignores biological/technical variations)
Input file format¶
# number before "," indicates number of methyl reads, and number after "," indicates
# number of total reads
cgID sample_1 sample_2
CpG_1 129,170 166,178
CpG_2 24,77 67,99
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Data file containing methylation proportions (represented by “methyl_count,total_count”, eg. “20,30”) with the 1st row containing sample IDs (must be unique) and the 1st column containing CpG positions or probe IDs (must be unique). This file can be a regular text file or compressed file (*.gz, *.bz2) or accessible url. -g GROUP_FILE, --group=GROUP_FILE Group file defining the biological groups of each sample. It is a comma-separated 2 columns file with the 1st column containing sample IDs, and the 2nd column containing group IDs. It must have a header row. Sample IDs should match to the “Data file”. -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Commands¶
$ dmc_fisher.py -i test_09.tsv.gz -g test_09.grp.csv -o test_fisher
Output¶
- 3 columns (“Odds ratio”, “pvalue” and “FDR adjusted pvalue”) will append to the original table.
$ head -5 test_fisher.pval.txt
ID LTS_MCR-1008 LTS_MCR-1035 STS_MCR-1021 STS_MCR-1251 OddsRatio pval adj.pval
chr10:100011340 12,14 26,37 0,18 10,24 9.353846153846154 1.2116597355208375e-06 6.343768248800197e-05
chr10:100011341 0,21 0,54 0,26 0,19 nan 1.0 1.0
chr10:100011387 0,14 0,40 0,20 0,24 nan 1.0 1.0
chr10:100011388 18,18 47,54 19,23 18,19 1.2548262548262548 0.7574366471769988 1.0
chr10:100026933 16,30 28,55 7,40 13,19 2.0926829268292684 0.04119183894184185 0.2617016451197068
dmc_glm.py¶
Description¶
This program performs differential CpG analysis using generalized liner model. It allows for covariants analysis.
Options¶
- Options:
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Data file containing beta values with the 1st row containing sample IDs (must be unique) and the 1st column containing CpG positions or probe IDs (must be unique). This file can be a regular text file or compressed file (.gz, .bz2). -g GROUP_FILE, --group=GROUP_FILE Group file defining the biological groups of each sample as well as other covariables such as gender, age. The first variable is grouping variable (must be categorical), all the other variables are considered as covariates (can be categorical or continuous). Sample IDs should match to the “Data file”. -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$dmc_glm.py -i test_05_TwoGroup.tsv.gz -g test_05_TwoGroup.grp.csv -o GLM_2G
$dmc_glm.py -i test_05_TwoGroup.tsv.gz -g test_05_TwoGroup.grp2.csv -o GLM_2G
Output files¶
- GLM_2G.results.txt
- GLM_2G.r
- GLM_2G.pval.txt (final results)
dmc_logit.py¶
Description¶
This program performs differential CpG analysis using logistic regression model based on proportion values. It allows for covariable analysis. Users can choose to use “binomial” or “quasibinomial” family to model the data. The quasibinomial family estimates an addition parameter indicating the amount of the overdispersion.
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Data file containing methylation proportions (represented by “methyl_count,total_count”, eg. “20,30”) with the 1st row containing sample IDs (must be unique) and the 1st column containing CpG positions or probe IDs (must be unique). This file can be a regular text file or compressed file (*.gz, *.bz2) or accessible url. -g GROUP_FILE, --group=GROUP_FILE Group file defining the biological groups of each sample as well as other covariables such as gender, age. The first variable is grouping variable (must be categorical), all the other variables are considered as covariates (can be categorical or continuous). Sample IDs should match to the “Data file”. -f FAMILY_FUNC, --family=FAMILY_FUNC Error distribution and link function to be used in the GLM model. Can be integer 1 or 2 with 1 = “quasibinomial” and 2 = “binomial”. Default=1. -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$ dmc_logit.py -i test_04_TwoGroup.tsv.gz -g test_04_TwoGroup.grp.csv -o output_quasibin
$ dmc_logit.py -i test_04_TwoGroup.tsv.gz -g test_04_TwoGroup.grp.csv -f 2 -o output_bin
dmc_nonparametric.py¶
Description¶
This program performs differential CpG analysis using the Mann-Whitney U test for two group comparison, and the Kruskal-Wallis H-test for multiple groups comparison.
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Data file containing beta values with the 1st row containing sample IDs (must be unique) and the 1st column containing CpG positions or probe IDs (must be unique). Except for the 1st row and 1st column, any non-numerical values will be considered as “missing values” and ignored. This file can be a regular text file or compressed file (.gz, .bz2). -g GROUP_FILE, --group=GROUP_FILE Group file defining the biological group of each sample. It is a comma-separated two columns file with the 1st column containing sample IDs, and the 2nd column containing group IDs. It must have a header row. Sample IDs should match to the “Data file”. Note: automatically switch to use Kruskal-Wallis H-test if more than two groups were defined in this file. -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
$dmc_nonparametric.py -i test_05_TwoGroup.tsv.gz -g test_05_TwoGroup.grp.csv -o U_test
$dmc_nonparametric.py -i test_06_TwoGroup.tsv.gz -g test_06_TwoGroup.grp.csv -o H_test
dmc_ttest.py¶
Description¶
Differential CpG analysis using T test for two groups comparison or ANOVA for multiple groups comparison.
Options¶
--version show program’s version number and exit -h, --help show this help message and exit -i INPUT_FILE, --input_file=INPUT_FILE Data file containing beta values with the 1st row containing sample IDs (must be unique) and the 1st column containing CpG positions or probe IDs (must be unique). Except for the 1st row and 1st column, any non-numerical values will be considered as “missing values” and ignored. This file can be a regular text file or compressed file (.gz, .bz2). -g GROUP_FILE, --group=GROUP_FILE Group file defining the biological group of each sample. It is a comma-separated 2 columns file with the 1st column containing sample IDs, and the 2nd column containing group IDs. It must have a header row. Sample IDs should match to the “Data file”. Note: automatically switch to use ANOVA if more than 2 groups were defined in this file. -p, --paired If True, performs a paired t-test (the paired sampels are matched by the order). If False, performs a standard independent 2 sample t-test. default=False -w, --welch If True, performs Welch’s t-test which does not assume the two samples have equal variance. If False, performs a standard two-sample t-test (i.e. assuming the two samples have equal variance). default=False -o OUT_FILE, --output=OUT_FILE The prefix of the output file.
Input files (examples)¶
Command¶
#Two group comparison. Compare normal livers to HCV-related cirrhosis livers
$dmc_ttest.py -i test_05_TwoGroup.tsv.gz -g test_05_TwoGroup.grp.csv -o ttest_2G
#Three group comparison. Compare normal livers, HCV-related cirrhosis livers, and liver cancers
$dmc_ttest.py -i test_06_ThreeGroup.tsv.gz -g test_06_ThreeGroup.grp.csv -o ttest_3G
Output files¶
- ttest_2G.pval.txt
- ttest_3G.pval.txt
Compare Differential CpG Analysis Tools¶
| Program | Input data | Method/Model | Co-variable |
| dmc_fisher.py | Proportion value (RRBS/WGBS) | Fisher’s exact test | No |
| dmc_logit.py | Proportion value (RRBS/WGBS) | Logistic regression (binom or quasi-binom) | Yes |
| dmc_bb.py | Proportion value (RRBS/WGBS) | Beta-binomial regression | Yes |
| dmc_ttest.py | Beta- or M-value (450K/850K) | Student’s T-test or ANOVA | No |
| dmc_glm.py | Beta- or M-value (450K/850K) | Generalized linear model | Yes |
| dmc_nonparametric.py | Beta- or M-value (450K/850K) | Mann–Whitney U test or Kruskal-Wallis H test | No |
| dmc_Bayes.py | Beta- or M-value (450K/850K) | Bayes estimation | No |
P-value distributions¶
Compare p-value distributions of dmc_ttest.py, dmc_glm.py, dmc_nonparametric.py (U test), and dmc_Bayes.py

Correlation of p-values of dmc_ttest.py, dmc_glm.py, dmc_nonparametric.py (U test), and dmc_Bayes.py

LICENSE¶
CpGtools is distributed under GNU General Public License (GPLv3)
This program is free software; you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation; either version 3 of the License, or (at your option) any later version. This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details. You should have received a copy of the GNU General Public License along with this program; if not, If not, see <https://www.gnu.org/licenses/>.
Reference¶
Wei T, Nie J, Larson NB, Ye Z, Eckel Passow JE, Robertson KD, Kocher JA, Wang L. CpGtools: A Python Package for DNA Methylation Analysis. Bioinformatics. 2019 Dec 6 Epub 2019 Dec 06